
医学界新聞プラス
[第4回]障害の出現場所からどの疾患を疑うか②
『末梢神経障害 解剖生理から診断,治療,リハビリテーションまで』より
連載 古賀道明
2022.09.30
末梢神経障害
解剖生理から診断,治療,リハビリテーションまで
Common diseaseでありながら,少なくない人が苦手意識を持つ末梢神経障害。そんな,とっつきにくくややこしい末梢神経障害をわかりやすく理解してもらうために,臨床に活きる知識を厳選して一冊にまとめました。
本書の特徴は,各末梢神経疾患にはどういう特徴があるかといったいわゆる教科書的な情報を網羅するとともに,「この症候が出たら疑うべき疾患は何か」「障害の出現場所から何の疾患を考えるか」「障害分布からどの疾患を導き出すか」といった,鑑別を行う視点からも末梢神経疾患をときほぐし,双方向から多角的に末梢神経障害をとらえるところにあります。そして,双方向からの診断アプローチに不可欠な解剖生理,生化学,神経病理も臨床に役立つ視点からたっぷり解説しています。
「医学界新聞プラス」では,本書の中から「末梢神経障害の種類」と「病歴聴取」という診断の基礎となるテーマを前半2回でお読みいただき,より具体的な診断の知識として「障害の出現場所からどの疾患を疑うか」を後半2回でご紹介します。
四肢近位部に障害が強い
四肢近位部に筋力低下が強い
1 病態
末梢神経障害でみられる四肢筋力低下は遠位部優位であることが圧倒的に多い.これは多くの末梢神経障害(特に多発ニューロパチー)は,長さ依存性の障害パターンを示し,起始部からより遠い部位(四肢遠位部)から障害されるためである.一方,神経根を病変の主座とする場合や,神経叢(腕神経叢,腰仙神経叢)病変では,長さ非依存性の障害をきたし,四肢近位筋に強い筋力低下をきたしうる.
2 原因疾患(表5‒3)
Guillain-Barré症候群(GBS) や慢性炎症性脱髄性多発(根)ニューロパチー(CIDP)は, 多発神経根障害と多発ニューロパチーの両者の臨床的特徴を有し,四肢近位部と遠位部が同等の筋力低下をきたす代表的な疾患である1).また,GBS と類似する臨床像を示す急性間欠性ポルフィリン症(AIP)も忘れてはならない.AIP は腹痛・嘔吐の数日~数週後に,急性進行性の四肢筋力低下をきたし,感覚障害はないかあっても軽く,脳脊髄液では蛋白細胞解離を示すなど,GBS と誤認されやすい2).AIP は四肢近位筋優位の筋力低下をきたしやすい一方,四肢筋力低下は左右非対称性で,自律神経症候(血圧変動や頻脈,消化管蠕動運動低下など)がGBS より高度にみられ,かつ再発しやすいなどの特徴を有する.さらに,神経根を障害の好発部位とする疾患として,サルコイドニューロパチーや神経リンパ腫症,癌性髄膜炎なども押さえておきたい.
神経叢病変でも四肢近位筋に強い筋力低下をきたしうる.神経痛性筋萎縮症は,片側のC5髄節支配筋を中心に手内筋までまだら状の筋力低下・筋萎縮をきたし,感覚障害はないかごく軽度であることが多い.腕神経叢が責任病巣と想定されている.糖尿病性腰仙神経叢根ニューロパチー(DLRPN) は,糖尿病性ニューロパチーの中でも左右非対称かつ局所性の障害をきたす主要な病型である3).下肢近位筋を障害することからproximal diabetic neuropathy と呼ばれることもある.背部や殿部,大腿部の疼痛で発症し,数日~数週後に一側下肢近位筋(殿部や大腿部)に進行性の筋力低下をきたし大腿部に筋萎縮を示すようになる.1/3 の症例では上肢近位筋にも筋力低下が拡大する.
遺伝性ニューロパチーの中でも四肢近位筋優位の筋力低下をきたす疾患として,近位筋優位遺伝性運動感覚性ニューロパチー(HMSN–P)がある.本疾患は,四肢の感覚障害や腱反射低下・消失に加え,四肢近位筋の筋力低下・筋萎縮を主徴とする.成人発症かつ緩徐進行性の常染色体優性遺伝疾患で,わが国では沖縄県と滋賀県に地域集積性がみられ,原因遺伝子としてTRK-fused が同定された4).神経病理所見として,末梢神経の有髄線維の高度脱落に加え,下位運動ニューロン優位の運動ニューロン疾患の病理像を呈し,末梢神経と運動ニューロンの両者の障害が,特徴的な臨床像の発現に関与している可能性が想定される5).

3 鑑別すべき疾患
各種筋疾患だけでなく,運動ニューロン疾患〔筋萎縮性側索硬化症(ALS) や脊髄性筋萎縮症(SMA),球脊髄性筋萎縮症(SBMA)〕が鑑別対象である.ALS は一般的に四肢遠位部の筋力低下で発症することが多いが,flail arm syndrome と呼ばれる病型(brachial amyotrophic diplegia とも呼ばれる)では両上肢の近位筋優位の筋力で発症するため6),鑑別対象となる.
四肢近位部・体幹に感覚障害が強い
原因疾患
後根神経節障害や単ニューロパチー,多発性単ニューロパチーをきたす疾患では,病変部位によって四肢近位部優位の感覚障害をきたす.AIP でも稀ながら,“bathing trunks”(男性用の水着で,大腿部の一部まで裾があるもの)様分布と表現される近位部の感覚障害がみられる1).
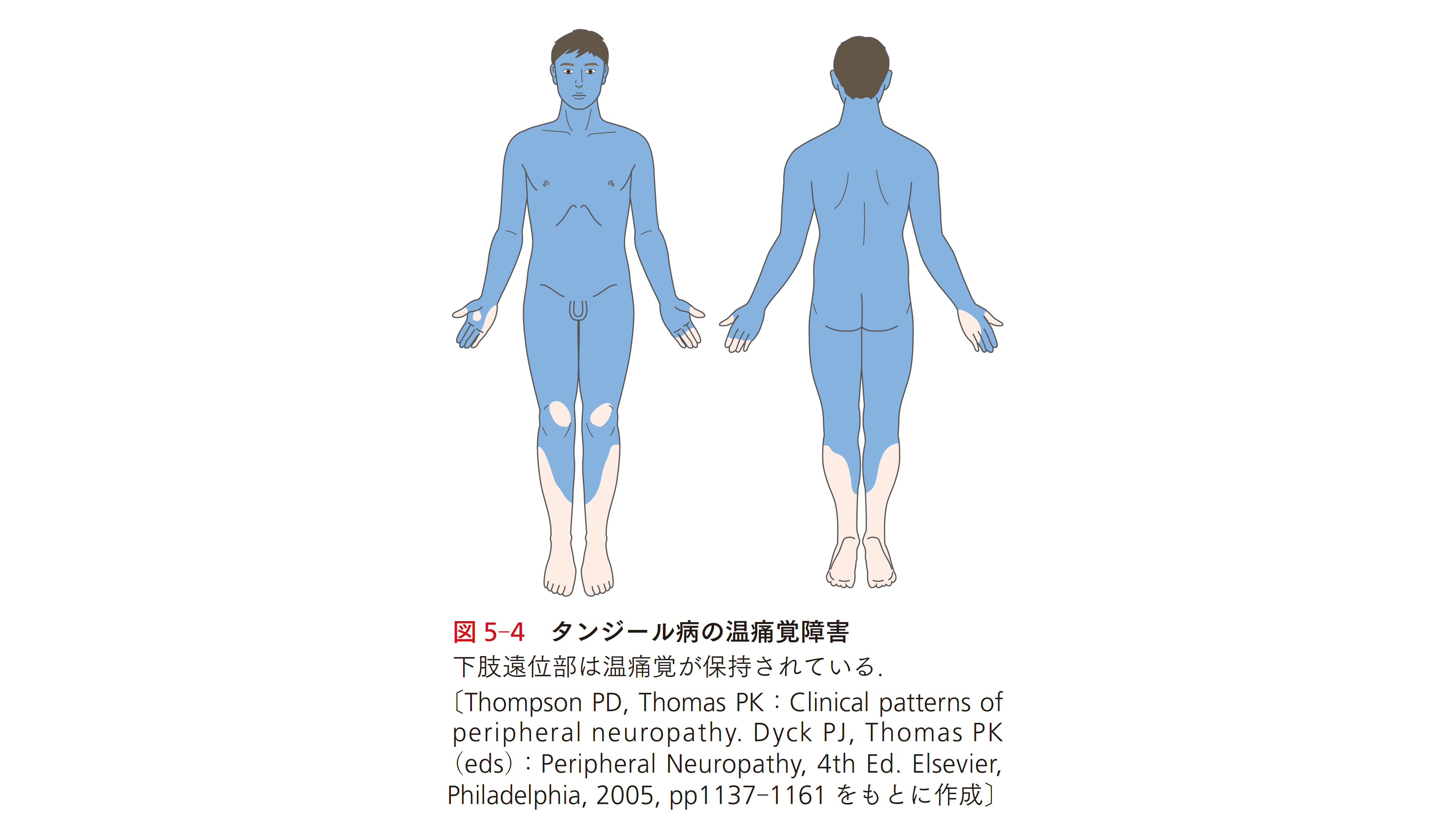
タンジール病は,コレステロール・トランスポーターであるATP– binding cassette transporter A1 の遺伝子異常で生じる常染色体劣性遺伝疾患である.HDL の欠損と種々の臓器にコレステロールエステルの蓄積をきたし全身の臓器障害がみられる.Schwann細胞にも脂質が沈着しさまざまなニューロパチーを呈すが,中でも「脊髄空洞症様ニューロパチー」と表現される病型が主要である7).この病型では,上肢や顔面を中心に筋力低下,筋萎縮をきたすのに加え,病初期には解離性感覚障害(温痛覚鈍麻かつ表在感覚は保持)を呈するが,下肢遠位部の感覚は保たれるのが特徴的である(図5‒4).なぜ,この特徴的な感覚障害分布をとるのかは明らかにされていない.神経症候以外には,HDL コレステロール値の高度低下やオレンジ扁桃(オレンジ色の腫大・分葉した扁桃),扁桃摘出術の既往,冠動脈疾患,肝脾腫大などの特徴があり,本症を疑うきっかけとなる.
脳神経障害が強い
脳神経が強く障害される原因
嗅神経(Ⅰ) と視神経(Ⅱ) 以外の脳神経(Ⅲ~Ⅻ)は,Schwann 細胞が髄鞘を形成し末梢神経に分類されるが,通常の末梢神経障害で脳神経が障害されることは多くない.これは末梢神経障害(特に多発ニューロパチー)の多くは,長さ依存性の障害パターンを示し,起始部からより遠い部位(つまり四肢遠位部)から障害されるため,四肢の末梢神経より短い脳神経は障害を免れやすいことによる.つまり,四肢の末梢神経よりも脳神経が強く障害される場合は,①脳神経を標的とする自己免疫性ニューロパチー,②多発性単ニューロパチー,③神経節障害(ガングリオノパチー,ニューロノパチー),④その他,に分けて考えると理解しやすい(表5‒4).

脳神経を標的とする自己免疫性ニューロパチー
GBS は四肢筋力低下を主徴とし,筋力低下が下肢→上肢→脳神経と上行性に拡大するのが典型的であるが,逆に脳神経麻痺(外眼筋麻痺など)で発症し,上肢→下肢と下行性に筋力低下が進展する症例が1 割程度存在する8).特にGBS の臨床亜型には脳神経障害を主徴とするものが多く,それらの半数以上の症例は四肢筋力低下をきたさない.先行感染の存在や脳脊髄液での蛋白細胞解離に加え,Miller Fisher 症候群(MFS)や咽頭頸部上腕型GBSではガングリオシド抗体が急性期血清中にほとんどの症例で検出されるため,診断に有用である.また,四肢に筋力低下がなくても腱反射の低下・消失や,神経伝導検査で四肢での伝導障害の存在が確認できれば,GBS の診断を支持する所見となる.
CIDP の約1 割の症例では,顔面神経麻痺や球麻痺などの脳神経障害を伴い,特に多巣性 CIDP〔以前は「multifocal acquired demyelinating sensory and motor (MADSAM)neuropathy」と呼ばれた〕でみられる脳神経障害は片側性であることが多い9).また,2021 年のCIDP の診療ガイドライン10)ではCIDP のカテゴリーから外されたものの,ノード・パラノード抗原に対する抗体〔コンタクチン1(CNTN1)抗体,コンタクチン関連蛋白質1(Caspr1)抗体,ニューロファシン(NF)140/186 抗体〕が陽性のニューロパチーでは,脳神経障害をきたしやすく注意が必要である11,12).
多発性単ニューロパチー
四肢にまったく障害がなく脳神経領域のみに障害がみられる場合,まずは脳神経神経核や脳神経の局所的な障害(脳血管障害や外傷,腫瘤,脳神経が通る神経孔・裂での圧迫など)の可能性につき,画像検査で鑑別することが必要である.
糖尿病性脳神経麻痺では四肢での多発ニューロパチーが顕在化する以前に出現することも多く,動眼神経や外転神経がそれぞれ単独かつ片側性に障害されることが多い.
水痘・帯状疱疹ウイルス(VZV)感染症では,患側耳介部から外耳道にかけての発疹と末梢性顔面神経麻痺を呈するRamsay Hunt 症候群が典型的である.しかし,発疹を欠き,顔面神経以外の脳神経が多発性に障害されることも多く,脳脊髄液検査で細胞数の増多があれば,神経サルコイドーシスや神経リンパ腫症などに加えVZV 感染症の可能性も想定したい.
Hansen 病はらい菌(Mycobacterium leprae)により四肢に感覚障害優位の多発性単ニューロパチーをきたすが,脳神経障害も2 割程度でみられ,顔面神経や嗅神経,三叉神経などが障害されやすい.
神経リンパ腫症は,非Hodgkin リンパ腫が末梢神経にびまん性に浸潤した状態であり,約2割の症例が脳神経障害で発症する12).
急性~亜急性の経過で両側性の顔面神経麻痺をきたす原因として,ライム病や神経サルコイドーシス,GBS などが代表的である.
神経節障害(ガングリオノパチー,ニューロノパチー)
末梢神経細胞体を標的とした自己免疫と考えられる病態で,三叉神経節細胞が障害されれば三叉神経領域の感覚障害をきたす.原因として,Sjögren 症候群,全身性エリテマトーデス(SLE),強皮症,混合性結合組織病などの膠原病が代表的である.
末梢神経細胞体を標的とした自己免疫と考えられる病態で,三叉神経節細胞が障害されれば三叉神経領域の感覚障害をきたす.原因として,Sjögren 症候群,全身性エリテマトーデス(SLE),強皮症,混合性結合組織病などの膠原病が代表的である.
その他
Charcot–Marie–Tooth 病(CMT)は,長さ依存性の多発ニューロパチーを呈し脳神経は保持されるのが一般的である.一方,SH3TC2 変異により発症する常染色体劣性遺伝性脱髄性ニューロパチーであるCMT タイプ4C(CMT4C)は,脳神経麻痺をきたしやすいことが特徴である.聴神経障害により聴力低下をきたすことが最も多く,脳神経麻痺以外には側弯症を伴いやすい13).そのほか,CMT の中で脳神経障害をきたすことが報告されている遺伝子異常として,PMP22(難聴をきたす)やMFN2(視神経萎縮),GDAP1(声帯麻痺),MPZ(Adie 瞳孔を伴う難聴)がある14).
わが国の遺伝性アミロイドニューロパチーは,トランスサイレチン型がほとんどで脳神経は保持される.一方,極めて稀ながらゲルソリン型(以前よりⅣ型やフィンランド型とも呼ばれる)では顔面神経麻痺を主体とする脳神経障害や角膜ジストロフィーを伴うのが特徴で15),わが国からも数家系の報告がある.
球症状が出現する
球症状を合併する原因
いずれの末梢神経障害でも高度となれば,原則として下位脳神経障害をきたし球症状を呈する.末梢神経障害が軽度にもかかわらず球症状を呈しやすい末梢神経障害は,①脳神経を標的とする自己免疫性ニューロパチー,②多発性単ニューロパチー,③咽頭など上気道に好発する感染症,④中枢神経障害を合併する(この場合は仮性球麻痺を呈する),⑤遺伝性末梢神経疾患,に分類することができる.①と②は前項で触れた.また,④は別章で紹介した疾患の多くが対象となる(本書第7章参照,本連載には掲載なし).以下では,①の中でもGBS と③,⑤に関して触れる.
GBS
球症状を主体とするGBS の臨床亜型として,急性口咽頭筋麻痺(筋力低下が口咽頭部に限局),咽頭頸部上腕型GBS(口咽頭部に加え,頸部,上腕の筋力低下),polyneuritis cranialis(外眼筋麻痺や顔面筋など脳神経が広範に障害される),Bickerstaff 脳幹脳炎(外眼筋麻痺・小脳性運動失調に加え意識障害などの中枢神経障害を伴う)があり,それぞれは連続した病態スペクトラムとみなすことができる16).球症状で発症するGBS の大部分でガングリオシド抗体(GT1a やGM1b,GQ1b などに対するIgG 抗体)が検出されるため,診断に有用である17).また,類似する神経症候を呈する疾患が末梢神経疾患以外にも多く,鑑別診断が重要となる18).
ジフテリア
球症状から発症し,脱髄性多発ニューロパチーを生じる原因としてジフテリアを押さえておきたい19).ジフテリアは,ジフテリア毒素を産生するCorynebacterium diphtheriaeによって生じる感染症で,わが国ではワクチンの定期接種が行われほとんどみられないが,輸入感染症として注意が必要である.咽頭や扁桃,喉頭などに感染した2~7 週間後に神経症候を発症し,初発症候として舌や顔面のしびれ感,発声障害が多い.嚥下障害や構音障害,呼吸器麻痺,外眼筋麻痺などに続き,毒素が血行性に全身に広がることで四肢麻痺に至る.一相性のGBS と異なり二相性の経過を示す.呼吸器ジフテリアの特徴として,咽頭・扁桃・喉頭の粘膜に灰白色の偽膜がみられ,本症を疑うきっかけとなる.
遺伝性疾患
Brown–Vialetto–Van Laere 症候群(BVVLS)は,小児期に発症し,感音性難聴や下位脳神経障害,呼吸不全を中核症候とする,常染色体劣性遺伝疾患である20).進行性の四肢筋力低下や球麻痺をきたすことから,ALS との鑑別が重要であるが,BVVLS では上位運動ニューロン徴候を示さず,また感覚性運動失調や腓腹神経生検で軸索脱落を認め,感覚性ニューロパチーとしての臨床的特徴を有する.原因遺伝子として,ビタミンB2として知られるリボフラビンのトランスポーター遺伝子(SLC52A3,SLC52A2)が同定され,リボフラビン高用量投与で一部の症例で症候の改善が報告されており,リボフラビンの欠乏が本疾患の原因となる可能性が想定されている.
慢性経過の感覚性運動失調型ニューロパチーに構音障害と外眼筋麻痺を合併するSANDO(sensory ataxic neuropathy, dysarthria, and ophthalmoplegia)症候群は,ミトコンドリア病の一病型で,慢性進行性外眼筋麻痺と同様のミトコンドリアDNA 異常を背景に発症すると考えられる21).
文献
1) Thompson PD, Thomas PK:Clinical patterns of peripheral neuropathy. Dyck PJ, Thomas PK(eds):Peripheral Neuropathy, 4th Ed. Elsevier, Philadelphia, 2005, pp1137–1161
2) Albers JW, Fink JK:Porphyric neuropathy. Muscle Nerve 30:410–422, 2004
3) Pasnoor M, Dimachkie MM, Barohn RJ:Diabetic neuropathy part 2:proximal and asymmetric phenotypes. Neurol Clin 31:447–462, 2013
4) Ishiura H, Sako W, Yoshida M, et al:The TRK-fused gene is mutated in hereditary motor and sensory neuropathy with proximal dominant involvement. Am J Hum Genet 91:320–329, 2012
5) 吉田眞理:近位筋優位遺伝性運動感覚ニューロパチー(HMSN‒P)の神経病理.臨床神経53:1200–1202, 2013
6) Jawdat O, Statland JM, Barohn RJ, et al:Amyotrophic lateral sclerosis regional variants(brachial amyotrophic diplegia, leg amyotrophic diplegia, and isolated bulbar amyotrophic lateral sclerosis). Neurol Clin 33:775–785, 2015
7) Mercan M, Yayla V, Altinay S, et al:Peripheral neuropathy in Tangier disease:A literature review and assessment. J Peripher Nerv Syst 23:88–98, 2018
8) Ropper AH, Wijdicks EFM, Truax BT:Guillain–Barré Syndrome. FA Davis, Philadelphia, 1991, pp73–105
9) Shibuya K, Tsuneyama A, Misawa S, et al:Cranial nerve involvement in typical and atypical chronic inflammatory demyelinating polyneuropathies. Eur J Neurol 27:2658–2661, 2020
10) Van den Bergh PYK, van Doorn PA, Hadden RDM, et al:European Academy of Neurology/Peripheral Nerve Society Guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy:Report of a joint Task Force‒ Second Revision. J Peripher
Nerv Syst 26:242–268, 2021
11) Delmont E, Brodovitch A, Kouton L, et al:Antibodies against the node of Ranvier:a real-life evaluation of incidence, clinical features and response to treatment based on a prospective analysis of 1500 sera. J Neurol 267:3664–3672, 2020
12) Baehring JM, Batchelor TT:Diagnosis and management of neurolymphomatosis. Cancer J 18:463–468, 2012
13) Piscosquito G, Saveri P, Magri S, et al:Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot–Marie–Tooth disease(CMT4). J Peripher Nerv Syst 21:142–149, 2016
14) Caress JB, Lewis JA, Pinyan CW, et al:A Charcot–Marie–Tooth type 1B kindred associated with hemifacial spasm and trigeminal neuralgia. Muscle Nerve 60:62–66, 2019
15) Kaku M, Berk JL:Neuropathy associated with systemic amyloidosis. Semin Neurol 39:578–588, 2019
16) Wakerley BR, Yuki N:Polyneuritis cranialis:oculopharyngeal subtype of Guillain–Barré syndrome. J Neurol 262:2001–2012, 2015
17) Koga M, Yuki N, Hirata K:Antiganglioside antibody in patients with Guillain–Barré syndrome who show bulbar palsy as an initial symptom. J Neurol Neurosurg Psychiatry 66:513–516, 1999
18) Ropper AH, Wijdicks EFM, Truax BT:Guillain–Barré Syndrome. FA Davis, Philadelphia, 1991, pp175–225
19) Pleasure D, Messing A:Diphtheritic polyneuropathy. Dyck PJ, Thomas PK(eds):Peripheral Neuropathy, 4th Ed. Elsevier, Philadelphia, 2005, pp2147–2151
20) Manole A, Fratta P, Houlden H:Recent advances in bulbar syndromes:genetic causes and disease mechanisms. Curr Opin Neurol 27: 506–514, 2014
21) Hanisch F, Kornhuber M, Alston CL, T et al:SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J Neurol Neurosurg Psychiatry 86:630–634, 2015

末梢神経障害
解剖生理から診断,治療,リハビリテーションまで
末梢神経障害の診断アプローチを双方向から徹底解説!
<内容紹介>末梢神経障害の臨床に必須の情報を網羅した、明日の診療がレベルアップする1冊。commonからrareまで重要な末梢神経疾患の特徴を幅広く解説することに加え、症候の種類・出現場所、どの神経に障害があるかといった所見から何を疑うべきかを解説し、双方向から疾患に迫る。双方向からのアプローチに欠かせない解剖生理、生化学、神経病理の“真に役立つ”知識を厳選。最新の治療、リハビリテーションまで充実の内容。
目次はこちらから
タグキーワード
いま話題の記事
-
忙しい研修医のためのAIツールを活用したタイパ・コスパ重視の文献検索・管理法
寄稿 2023.09.11
-
人工呼吸器の使いかた(2) 初期設定と人工呼吸器モード(大野博司)
連載 2010.11.08
-
連載 2010.09.06
-
事例で学ぶくすりの落とし穴
[第7回] 薬物血中濃度モニタリングのタイミング連載 2021.01.25
-
寄稿 2016.03.07
最新の記事
-
医学界新聞プラス
[第1回]心エコーレポートの見方をざっくり教えてください
『循環器病棟の業務が全然わからないので、うし先生に聞いてみた。』より連載 2024.04.26
-
医学界新聞プラス
[第1回]バルーン閉塞下逆行性経静脈的塞栓術(BRTO)
『IVRマニュアル 第3版』より2024.04.26
-
医学界新聞プラス
[第4回]脆弱性骨盤骨折
『クリニカル・クエスチョンで考える外傷整形外科ケーススタディ』より連載 2024.04.26
-
医学界新聞プラス
[第3回]わかりやすく2つの軸で分類して考えてみましょう
『心理社会的プログラムガイドブック』より連載 2024.04.26
-
医学界新聞プラス
[第1回]平坦な病変 (1)色調の変化があるもの
『内視鏡所見のよみ方と鑑別診断——上部消化管 第3版』より連載 2024.04.26
開く
医学書院IDの登録設定により、
更新通知をメールで受け取れます。
医学界新聞公式SNS
シェアする



URLをコピーしました