
- 医学
新年号特集 医薬品開発の未来を展望する カラー解説
創薬における日本の現状と国際動向
寄稿 土井 俊彦
2025.01.14 医学界新聞:第3569号より
シェアする
医薬品開発,創薬を行うには,基礎研究から実用化に至るまでの幅広い研究開発能力とともに,社会制度や規制等の構築も含めた国としての総合力が必要である。この総合力を涵養するため,創薬競争の世界でエコシステムを構成する人材,関連産業,臨床基盤に加えて,国の産業支援を最適化し,国際的な視点を踏まえながら,現実的な対策を講じていくことが不可欠となる。近年の新規医薬品開発は複雑化していることから,基礎研究,臨床試験,製造,販売までの一連のプロセスを製薬企業が単独で担う体制から脱却し,新興バイオベンチャーを中心にアカデミア・製薬企業・国といった各ステークホルダーが共同体を形成しなければならない。そうでなければ,高度化したシーズ(医薬品の元となる物質)開発を進められないであろう。人材や資金といったリソースがつながり,最適化された創薬エコシステムが実現した先で,日本がグローバル創薬エコシステムの一翼を担うことが理想的である。
医薬品開発のトレンド変化と取り残される日本
過去,日本は創薬先進国として画期的医薬品(ファースト・イン・クラス新薬)を創出していたが,今やその地位は低下した。日本が得意としていた,従来の低分子モダリティを中心とした創薬にビジネス的な限界が訪れたためだ。海外のメガファーマは組織再編や事業転換を行い,がんや難病・希少領域疾患薬の開発へと舵をきった。がん領域での分子標的治療薬やバイオマーカー主導の個別化医療は成功例も認めたが,その結果患者数が少なく開発コストの回収が難しいオーファンドラッグ化が進んだ。また,バイオテクノロジーの発達によって武装化抗体,再生細胞医薬などの新しい医薬品ビジネスが生み出され,それらの分野でユニバーサル医薬を作り出そうとしている。そうした新しいスタイルの創薬では,基礎研究・開発研究・製品化・市場化を一貫して行う開発基盤が必要となる。一方で早期からの成功率は高くはないため,投資コストが莫大になる。結果,バイオテクノロジー医薬品は高額となる傾向が強く,対象となる疾患が増えることでコストパフォーマンスが良くなる創薬エコシステムが注目されることになった。加えてここ数年間,COVID-19ワクチンに代表される遺伝子創薬やRNA創薬が注目され,グローバル治験やデジタルトランスフォーメーション,DCT(分散化臨床試験:来院に依存しない臨床試験手法)など,臨床試験・開発手法も大きく変わりつつある。
日本の製薬企業が自らの創薬力を低下させた結果,海外からのライセンシングに依存する事態を招いた。海外の製薬企業では研究機能をアウトソーシングし,最先端の技術を持つバイオベンチャーを買収することで新薬候補拡充を行う効率化とリスク分散化を進めている。結果的にバイオベンチャーの分業・多様化が進んでおり,ベンチャー設立の目的も自ら株式市場に上場するIPOから,メガファーマに事業を売却するM&Aへとシフトした。日本では,アカデミア発のシーズ導出を目的にベンチャーが設立されてはいるが,出口戦略は国内でのIPOに偏っているのが現状である。日本の創薬環境は世界の中で取り残されているのが現状であり,人材教育,トレーニングの場もないため人材が育たない。創薬エコシステム構築のためにも,他国での経験・ノウハウを有する人材を呼び込む必要があるだろう(図1)。
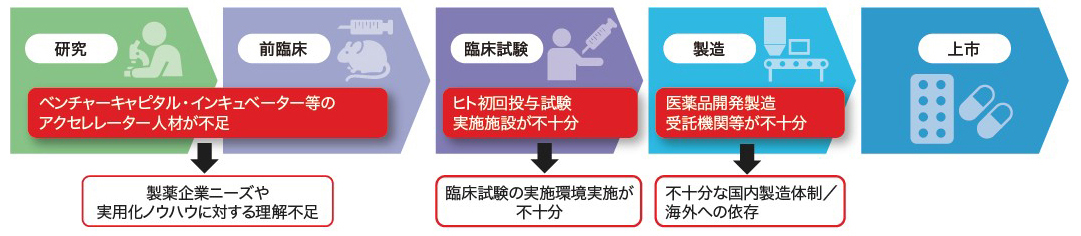
人材の流動性が乏しいこともあり,製薬企業ニーズや実用化ノウハウを熟知した人材がアカデミアやスタートアップに不在となっている。そのため,早期段階から実用化を見据えた研究が十分に実施されていない。エコシステムの実現に向けて,米国等における実用化ノウハウを有する人材を呼び込む必要がある。外資系メガファーマや米国系VCからの人材・投資の誘致も検討する余地がある。
深刻化するドラッグ・ラグ/ロス問題とその原因
海外で開発された新薬の承認が遅れる「ドラッグ・ラグ」,そもそも新薬が国内に入ってこない「ドラッグ・ロス」が日本では深刻化している。2014年以降に欧米で承認・上市された製品のうち,既存モダリティで27%,新規モダリティで35%がドラッグ・ロス状態にある(図2,座談会記事の表参照)。この傾向は今後より深刻になる見通しだ(座談会記事の図1参照)。
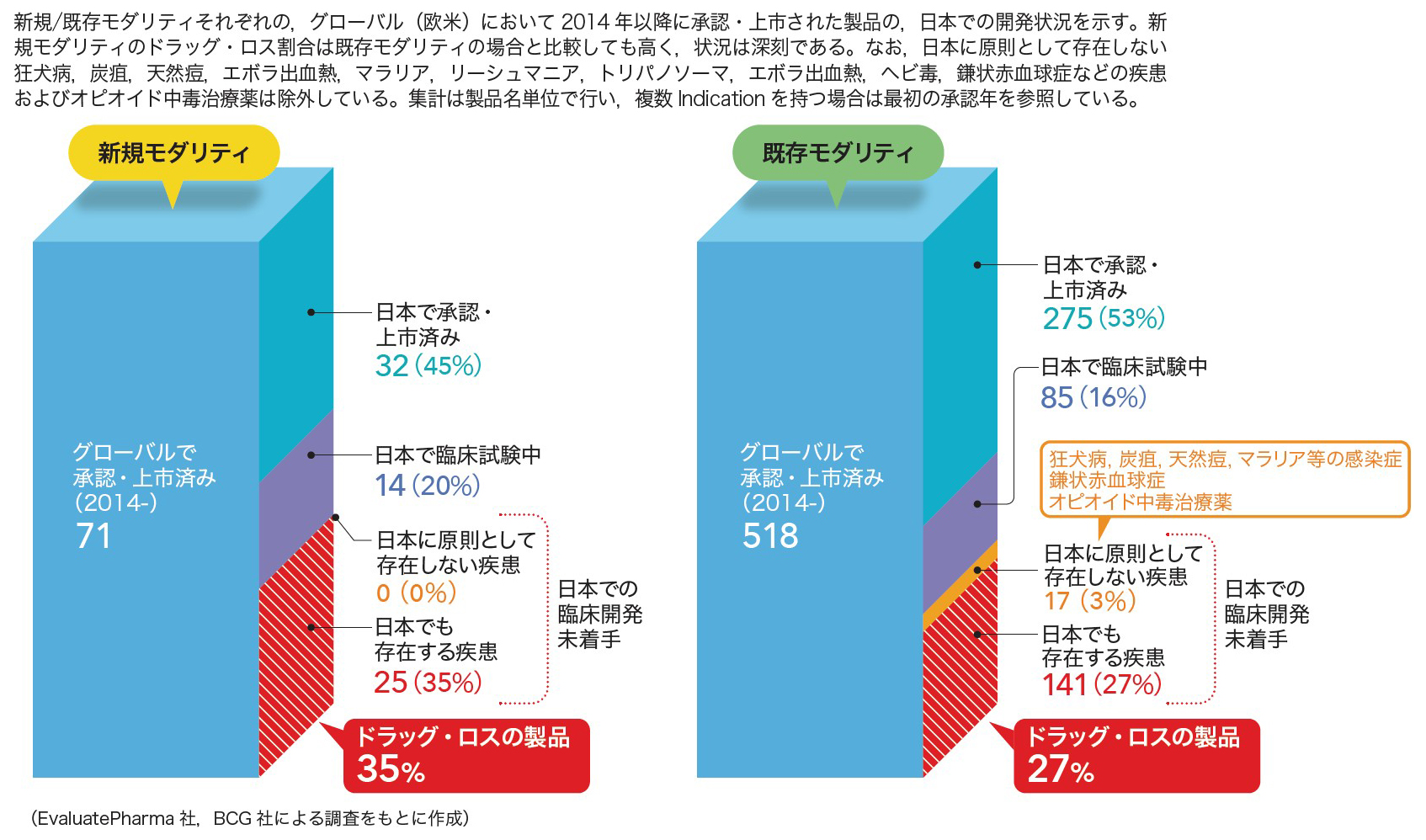
このようなドラッグ・ラグ/ロス問題の原因として,治験・承認プロセスの在り方が挙げられる。具体的には審査期間が長い,体制の違いから国際共同治験に日本を組み入れにくい,薬事・薬価制度の透明性・予見性が低い(特許期間中の薬価引き下げなど)というようにさまざまな原因分析がなされている。特にドラッグ・ラグ/ロス問題が目立つがん領域は行政問題とも言われているが,患者数の多いがん領域での承認が海外に比して遅いわけではないなど,広い視野での冷静な現状分析も必要だろう。
こうした問題に対し,日本もただ手をこまねいていたわけではない。①医薬品医療機器総合機構(PMDA)審査官の増強,②国際共同治験への参加推奨〔「国際共同治験に関する基本的考え方」(2007年)〕,③日本で国際誕生日(註1)を迎えた場合の薬価インセンティブ〔「新薬創出・適応外薬解消等促進加算(新薬創出加算)」(2010年)〕,④ドラッグ・ラグ/ロスへの直接的対応〔「医療上の必要性の高い未承認薬・適応外薬検討会議」(2010年~)〕,⑤海外治験データの有効利用といった取り組みが行われている。臨床試験・研究・治験基盤の体制整備についても,臨床研究中核病院を位置づけ,治験体制整備による国際共同治験や医師主導治験,早期臨床試験(第Ⅰ相治験)への参加の推進などが進んできた。その結果,ドラッグ・ラグについては改善を認め,特定のがん領域では日本が世界をリードする可能性も見えてきている。
最近新たに生じているドラッグ・ラグ/ロスの原因は,明らかに様相が異なる。先ほど述べたように現在の開発ターゲットは希少疾患が中心であり,その開発はベンチャーが担う。そうした...
この記事はログインすると全文を読むことができます。
医学書院IDをお持ちでない方は医学書院IDを取得(無料)ください。
土井 俊彦 国立がん研究センター東病院 病院長
いま話題の記事
-
ピットフォールにハマらないER診療の勘どころ
[第22回] 高カリウム血症を制するための4つのMission連載 2024.03.11
-
VExUS:輸液耐性が注目される今だからこそ一歩先のPOCUSを
寄稿 2025.05.13
-
サルコペニアの予防・早期介入をめざして
AWGS2025が示す新基準と現場での実践アプローチ寄稿 2026.03.10
-
寄稿 2022.04.11
-
医学界新聞プラス
[第4回]セフェム系抗菌薬 / 臓器別感染症:市中肺炎
これで身につく! 感染症まるごとスタートダッシュ連載 2026.02.05
最新の記事
-
どう届ける? いかに選ばれる?
with AI 医療コミュニケーション対談・座談会 2026.07.14
-
対談・座談会 2026.07.14
-
EBPを生きた実践にする
基礎教育からDNPまで,求められる学びと役割対談・座談会 2026.07.14
-
インタビュー 2026.07.14
-
藤澤雄太氏に聞く
患者と信頼関係を築く動機づけ面接
説得ではなく伴走で変化を促すインタビュー 2026.07.14
開く
医学書院IDの登録設定により、
更新通知をメールで受け取れます。